Elucidating 5-HT2A's true antidepressant/neuroplasticity pathway

Purpose of this post
There hasn’t been any review article that put all the pathways (G-proteins and β-arrestin) together and helping understand which pathway actually does what, like contributing to hallucinations (HTR) or antidepressant effects/neuroplasticity.
It’s not even well known yet how psychedelics truly cause hallucinations (measured by HTR in mice) or which pathway is actually necessary for antidepressant effects, this post is for the antidepressant pathway.
From my research, the three major clues that solidify the entire antidepressant theory, leaving only small things to be safely/easily elucidated with strong plausibility.
Confirming intracellular 5-HT2A produces the neuroplasticity/antidepressant effects and extracellular 5-HT2A doesn’t.
This is the only mechanism that’s well-known by researchers and referenced a lot, because it became one of the most popular psychedelic papers.
Fully biased β-arrestin non-hallucinogenic psychedelics being able to produce the full antidepressant effect, indicating Gq-protein isn’t necessary.
The β-arrestin pathway isn’t associated with hallucinations at all in psychedelics.
Confirming psychedelics don’t use the β-arrestin/PI3K/Akt complex, but instead the β-arrestin travels to the nucleus (Class A), then assembles the β-arrestin/C-Raf/MEK/ERK complex.
This review is the only 5-HT2A review even attempting to show all the missing mechanisms of how psychedelics produce neuroplasticity/antidepressant effects.
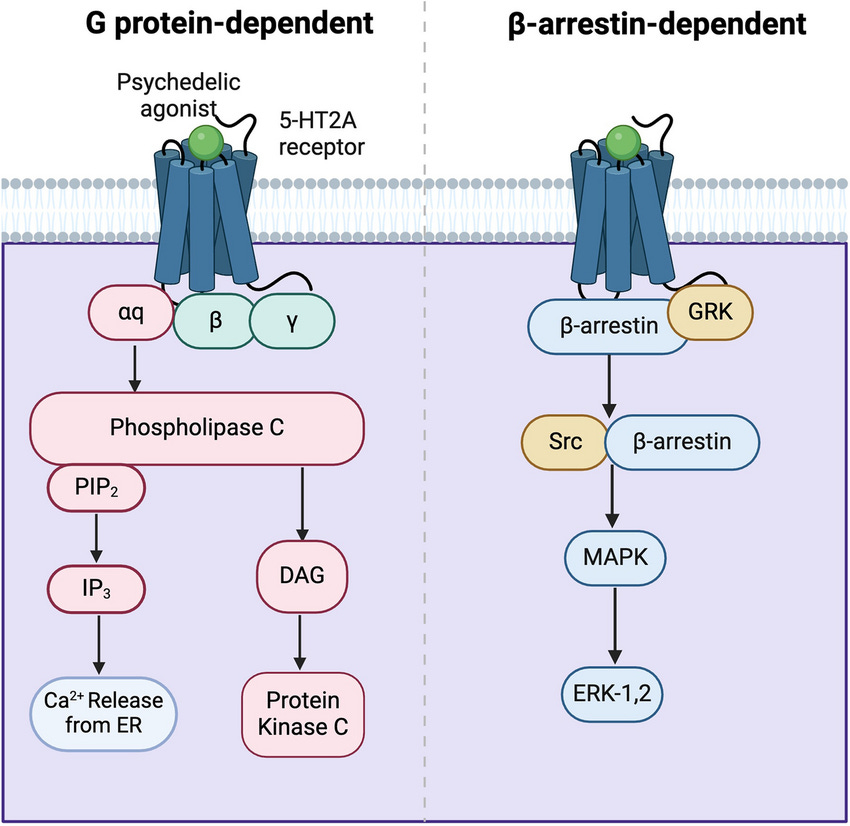
Antidepressant effects pathways: Gq-protein, β-arrestin, & intracellular 5-HT2A
Intracellular 5-HT2A: Colocalization endomembrane system
Starting the review off with intracellular 5-HT2A since it makes the difference between having neuroplasticity/antidepressant effects and not compared to extracellular 5-HT2A.
Extracellular 5-HT2A are predominantly located on the proximal apical dendrites of pyramidal neurons, which is too far away from where the endomembrane system (golgi, lysosomes, and nucleus together) is located which is in the center of the cell body [x, x, x].
Extracellular 5-HT2A are predominantly located on the apical dendrites
Whereas intracellular 5-HT2A are predominantly located on the golgi, the nucleus contains the DNA for gene transcription, and mTORC1 resides on lysosomes, making the endomembrane system’s compartments colocalized with intracellular 5-HT2A.
That’s a simple explanation as to why an intracellular receptor located in the endomembrane system has far greater potential for protein synthesis than an extracellular receptor.
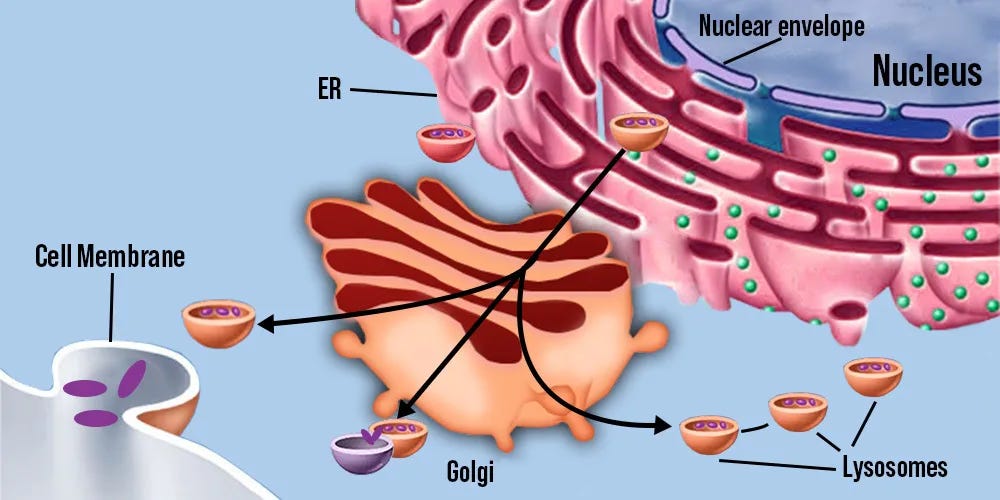
Endomembrane system compartments
The intracellular 5-HT2A paper (Vargas et al., 2023) remains the best evidence that intracellular 5-HT2A is what produces significant neuroplasticity and is linked to antidepressant effects, but not extracellular 5-HT2A, shown by the study’s extensive experiments using process of elimination.
This explains why Serotonin doesn’t produce significant neuroplasticity, because it’s neuronally impermeable, so cannot access intracellular 5-HT2A.
I summarized each in vivo/in vitro experiments at the very bottom of this post if you want to check it out, because it’s too big to add in this section.
In vitro and in vivo experiments to prove intracellular 5-HT2A is necessary and extracellular 5-HT2A is not for neuroplasticity and antidepressant effects
Extracellular 5-HT2A isn’t necessary, and that neuronal permeability to bypass the neuron’s membrane is how to access intracellular 5-HT2A, explaining why [x].
This paper is the proof that hallucinations aren’t necessary for antidepressant effects, because the 5-HT2A - mGluR2 heterodimer, which is necessary for hallucinations/HTR are extracellular.
mGluR5 is another example of an intracellular receptor that’s far more well researched than intracellular 5-HT2A that can be used as a comparison to see what’s most likely happening with the insufficiently researched intracellular 5-HT2A.
Note that intracellular mGluR5 is uniquely associated with LTD, whereas extracellular mGluR5 is associated with LTP, but we can still see how an intracellular receptor’s colocalization with the endomembrane system changes transcription efficacy significantly [x].
Intracellular mGluR5 does indeed have extended signaling because of the golgi’s acidity, like what’s assumed to happen with intracellular 5-HT2A [x, x].
mGluR5 has been found to be able to move between the golgi and ER (endoplasmic reticulum), back and forth, and when agoniszd by Glutamate, can activate signaling in these intracellular compartments [x, x].
Interestingly, for mGluR5, β-arrestin 2 is necessary for ERK activation and protein synthesis, whereas the Gq-protein pathway isn’t [x].
Sadly, the authors didn’t investigate the difference between intracellular and extracellular mGluR5, because it was found that fully biased β-arrestin 2 5-HT2A agonists are sufficient for antidepressant effects.
Additionally, extracellular mGluR5 only activates CREB, whereas intracellular mGluR5 activates both CREB and Elk-1, meaning better at inducing a greater amount of genes expressed [x].
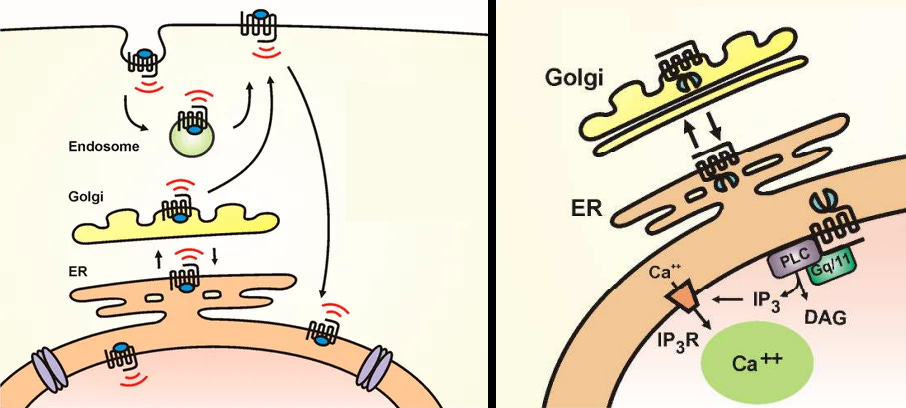
Known intracellular mGluR5 interactions in the endomembrane system that may apply to intracellular 5-HT2A
Interestingly, the inside of the golgi is acidic which allows for extended duration of G-protein coupled receptor signaling, but the inside of the ER isn’t acidic.
It’s been theorized that the acidity of the golgi can protonate psychedelics and lead to extended signaling by the authors of the intracellular 5-HT2A paper (Vargas et al., 2023), but I don’t believe this is true.
“A substantial proportion of 5-HT2ARs in cortical neurons are localized to the Golgi, and intracellular compartments such as the Golgi are slightly acidic compared with the cytosol and extracellular space.
Thus, it is possible that protonation of psychedelics within the Golgi leads to retention and sustained signaling, which results in neuronal growth, even after transient stimulation.”
There’s a big contradiction to their theory; the cytosol has too neutral of a pH to protonate psychedelics and the psychedelics aren’t actually inside the golgi when bound to intracellular 5-HT2A, they are still in the cytosol as the agonist site part of the receptor is in the cytosol and the signaling part is in the golgi’s enclosed space (lumen).
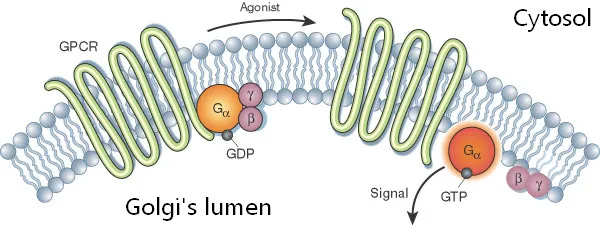
5-HT2A receptor (squiggly green) is partly outside the golgi which is in the cytosol, where the psychedelic binds
The receptor’s signaling part (orange/purple circles) inside the golgi’s lumen.
A much more likely theory is that since other G-protein coupled receptors (GPR4/65/68) in local acidic microenvironments can have their amino acid residues protonated, leading to extended signaling, this likely applies to intracellular 5-HT2A at the acidic golgi [x].
In summary, when looking at intracellular mGluR5’s interaction, it has long lasting signaling because of the acidity, drives transcription of a wider set of genes (CREB, Elk-1), and β-arrestin 2 may be significantly better at transcription than Gq-protein by measuring by ERK activation and protein synthesis.
Some or all of these intracellular mGluR5 mechanisms may apply to intracellular 5-HT2A because they both colocalize with the endomembrane system.
An interesting similarity is that mGluR5’s β-arrestin 2 pathway is necessary for ERK and protein synthesis, but not its Gq-protein pathway, implying being better nuclear ERK activation, similar to how 5-HT2A’s β-arrestin 2 pathway is completely sufficient for full antidepressant effects.
Colocalization with the endomembrane system is necessary because extracellular 5-HT2A isn’t associated with significant neuroplasticity/antidepressant effects.
Gq-protein: Not necessary for antidepressant effects, but can assist and is important for normal cognitive function
To start off, Gq-protein isn’t necessary for antidepressant effects, as the fully β-arrestin biased non-hallucinogenic psychedelics with zero Gq-protein efficacy (IHCH-7086, IHCH-7079) produced antidepressant effects matching LSD [x].
Only the Gq/s/i-protein pathways are associated with hallucinations in psychedelics, which is why IHCH-7086 and IHCH-7079 are non-hallucinogenic, full details in my other review.
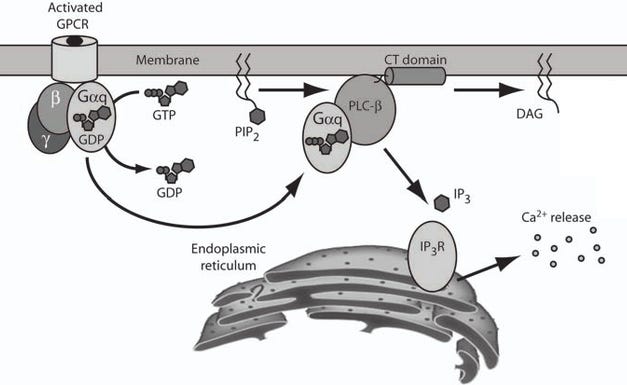
How the 5-HT2A Gq-protein (Gαq) pathway releases Ca2+ stored inside the ER (endoplasmic reticulum) out to the cytosol, the Ca2+ then activates PKC and CaM pathways
Still, there's a bunch of quite useful pathways associated with Gq-protein and intracellular 5-HT2A are located in an acidic environment that allows for sustained Gq-protein signaling.
These examples are below.
PKC/ERK pathway downregulates HDAC2, allowing more access to neuroplasticity/antidepressant associated genes [x].
HDAC2 is the main repressor of neuroplasticity/antidepressant genes, so downregulating HDAC2 makes gene transcription significantly easier [x].
Reducing HDAC2 is important for sustaining normal mGluR2 expression, because the mGluR2 gene promoter is repressed by HDAC2 [x].
5-HT2A antagonism (Clozapine) results in downregulation of mGluR2, because it prevents 5-HT2A from downregulating HDAC2 [x].
High NF-κB is neuroinflammatory and the PKC/ERK inhibits it [x].
Seems to synergistically activate ERK with β-arrestin, my theory is below [x].
PKCδ and CaMKII are both associated gene transcription by being able to activate ERK or CREB [x, x].
PKC directly activates the C-Raf
>MEK>ERK cascade.Associated with immediate early genes (c-Fos) [x].
CaMKII/IV phosphorylates CREB (Ser133) to activate it and CaMKII/IV also inhibits phosphatases that can deactivate CREB, resulting in longer lasting CREB transcription [x, x].
CaMKIV has short-lasting CREB activation (Ser133), whereas ERK activation has a sustained CREB activation (Ser133) [x].
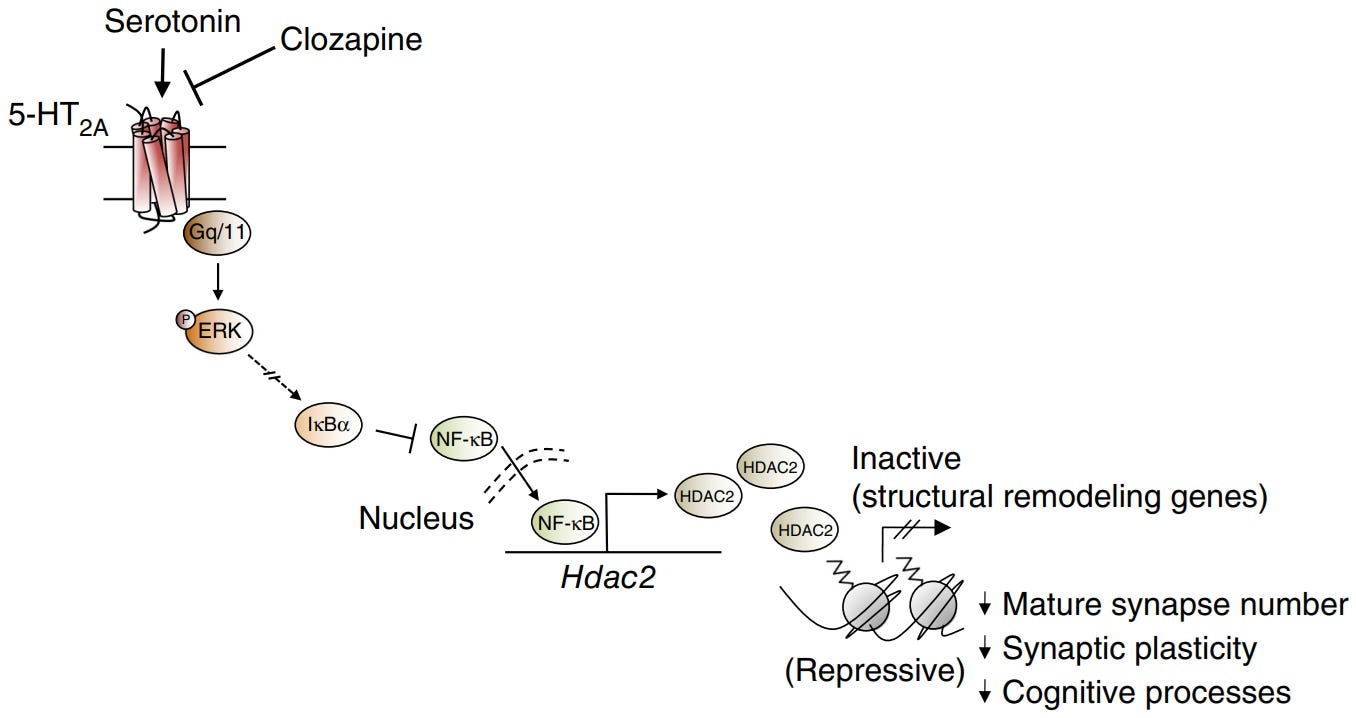
5-HT2A Gq-protein/PKC/ERK pathway downregulates HDAC2 by inhibiting NF-κB, since NF-κB upregulates HDAC2
Note that hallucinogenic psychedelics, which have high Gq-protein efficacy, can uniquely induce immediate early gene expression (c-Fos, Egr-1/2), whereas non-hallucinogenic psychedelics can’t.
But evidently these immediate early genes unique to hallucinogenic psychedelics have minimal/negligible contribution to neuroplasticity/antidepressant effects, details in my other review.
Those are the few mechanisms of the many ways the 5-HT2A Gq-protein pathway assists in enhancing gene transcription/translation, even though Gq-protein isn’t technically necessary for antidepressant effects.
Additionally, without going into much detail since it’s not on topic for depression, the 5-HT2A Gq-protein pathway is too important for normal cognitive function, it’s not a good idea to get rid of the Gq-protein pathway.
The reason the Gq-protein pathway’s ERK pathway is unsurprisingly not required for a psychedelic to produce full antidepressant effects is because it uses Ras/C-Raf, discussed in detail below.
Sustained nuclear ERK activity is required for significant neuroplasticity: Gq-protein versus β-arrestin
As previously mentioned, Gq-protein isn’t “necessary” for antidepressant effects, as the fully β-arrestin biased non-hallucinogenic psychedelics with zero Gq-protein efficacy (IHCH-7086, IHCH-7079) produced antidepressant effects matching LSD’s efficacy [x].
The key takeaway is that 5-HT2A’s β-arrestin pathway alone is sufficient to match the full antidepressant effects of a typical psychedelic like LSD.
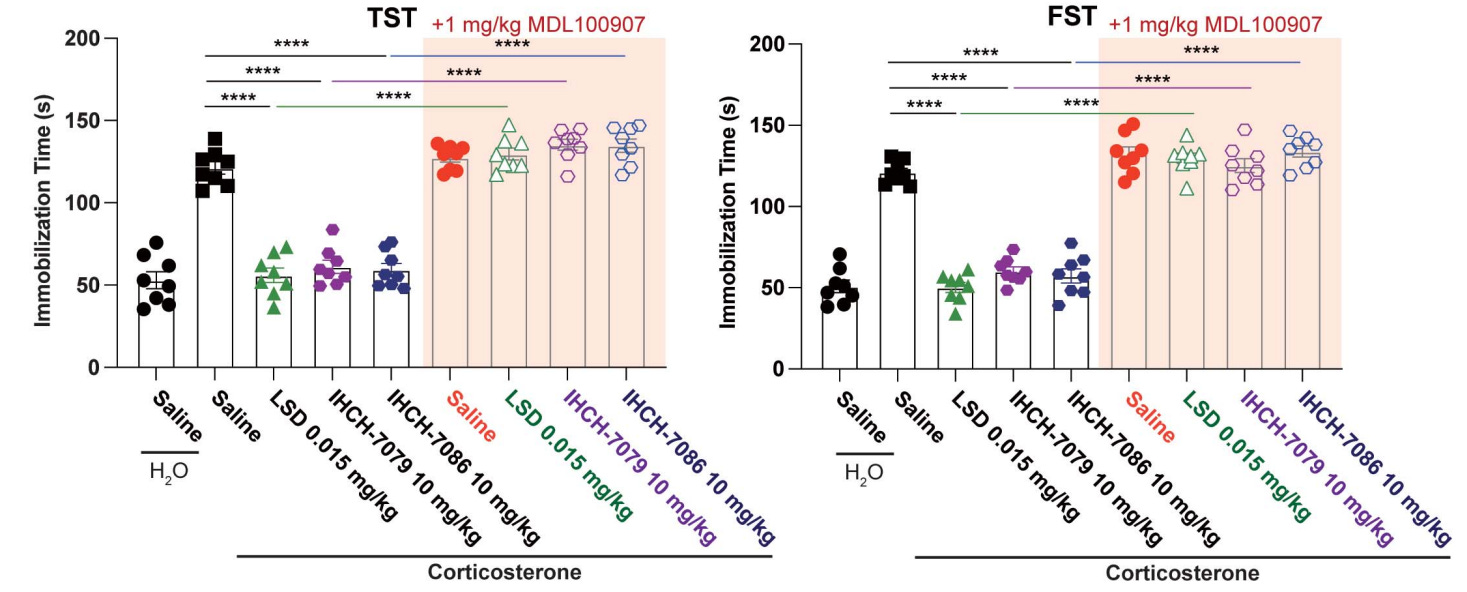
IHCH-7079 (purple circle) and IHCH-7086 (blue circle) compared to LSD (green triangle) antidepressant effects
To compare the neuroplasticity potential of Gq-protein versus β-arrestin, we need to compare their ability to activate ERK, because ERK activity is what drives gene transcription, leading to the synthesis of proteins that are required for neuroplasticity like BDNF.
It’s an important ERK location distinction between the nucleus and cytosol, since ERK must be inside the nucleus to drive transcription, because the nucleus is where DNA and the transcription factors like CREB are located [x, x, x].
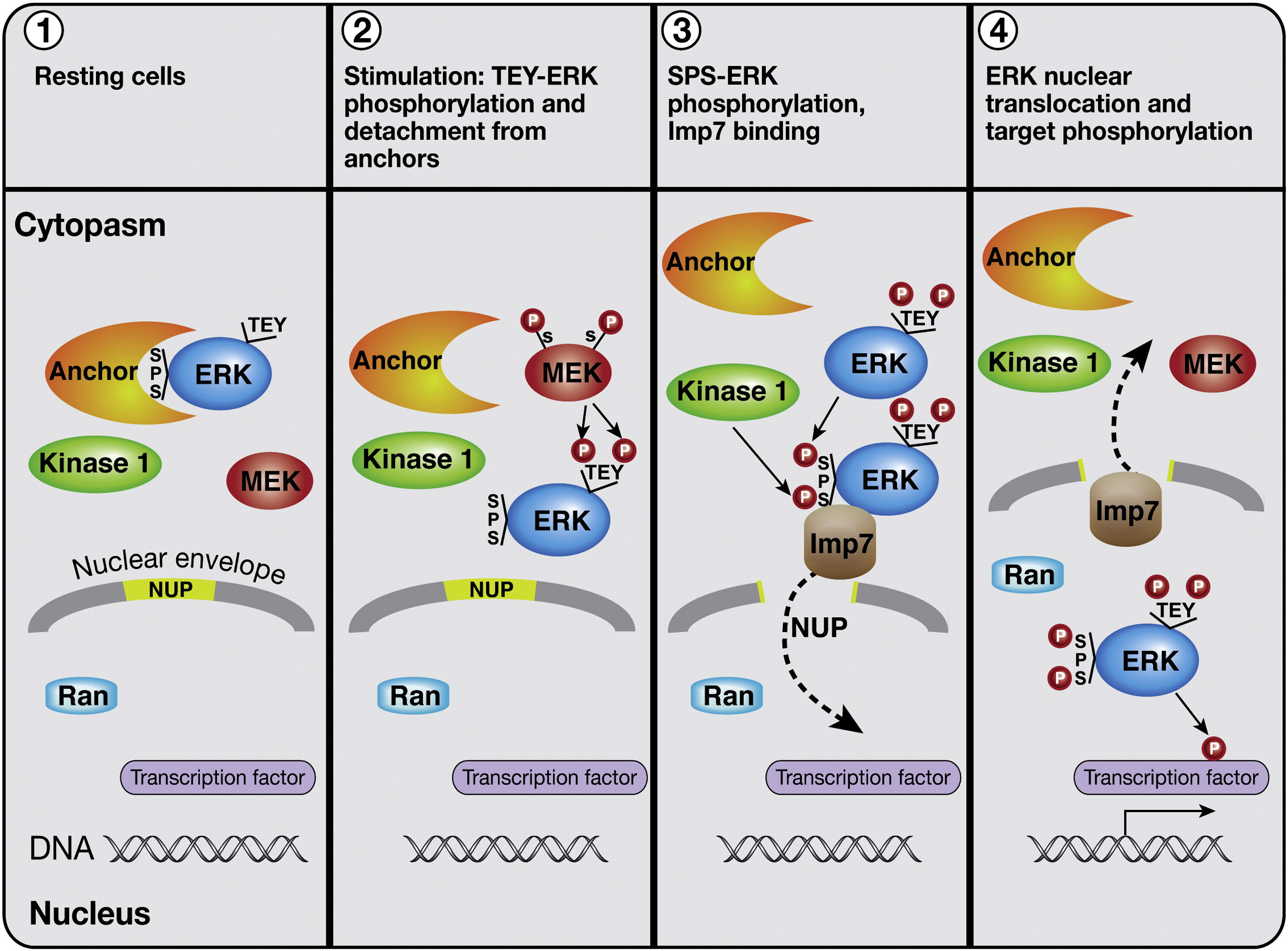
Nuclear ERK stabilizes the DNA’s transcription factors, which then drives transcription of proteins associated with neuroplasticity
But nuclear ERK isn’t enough, it must be sustained ERK activity.
C-Raf has a “short burst of ERK activity,” it gets inside the nucleus, but then is quickly cleared out the nucleus after the “short burst” of ERK is over, which is insufficient time to stabilize transcription factors to commit to significant transcription.
B-Raf has “sustained ERK activity,” it provides a constant flood of ERK to the nucleus that remains above the nuclear export rate, so nuclear ERK accumulates long enough to stabilize transcription factors, driving significant transcription [x, x, x, x, x, x, x].
This is because C-Raf is quickly terminated, whereas B-Raf is resistant to being turned off.
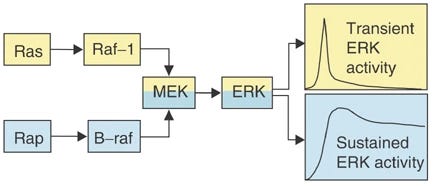
B-Raf has sustained ERK activity
C-Raf (Raf-1) is only good for strong/peak ERK activity, but is quickly terminated
So in terms of total gene transcription, sustained ERK activity easily wins.
The 5-HT2A Gq-protein pathway uses C-Raf, so ERK activity is too short and lacks potential to produce significant neuroplasticity.
It’s not surprising that the Gq-protein pathway isn’t necessary at all for the full antidepressant effects of psychedelics.
I know I also mentioned that intracellular 5-HT2A has sustained signaling earlier because of the golgi’s acidity, which would apply to Gq-protein.
But β-arrestin (late phase signaling) does end up blocking G-protein (early phase signaling) after some time, so intracellular 5-HT2A’s Gq-protein still shouldn’t be able to provide sufficient sustained ERK activity.
TrkB uses B-Raf and is known to be required for neuroplasticity and antidepressant effects in neuroplastogens.
5-HT2A
>Gq-protein>PKC>Ras>C-Raf>MEK>short lasting ERK activityTrkB
>Rap1>B-Raf>MEK>sustained ERK activity
To highlight the importance of sustained ERK activity; increasing the duration of sustained ERK activity with a DUSP6 inhibitor (BCI), results in significantly increased neuroplasticity and antidepressant effects extended from 1 week to 8 weeks in a neuroplastogen (Ketamine) [x].
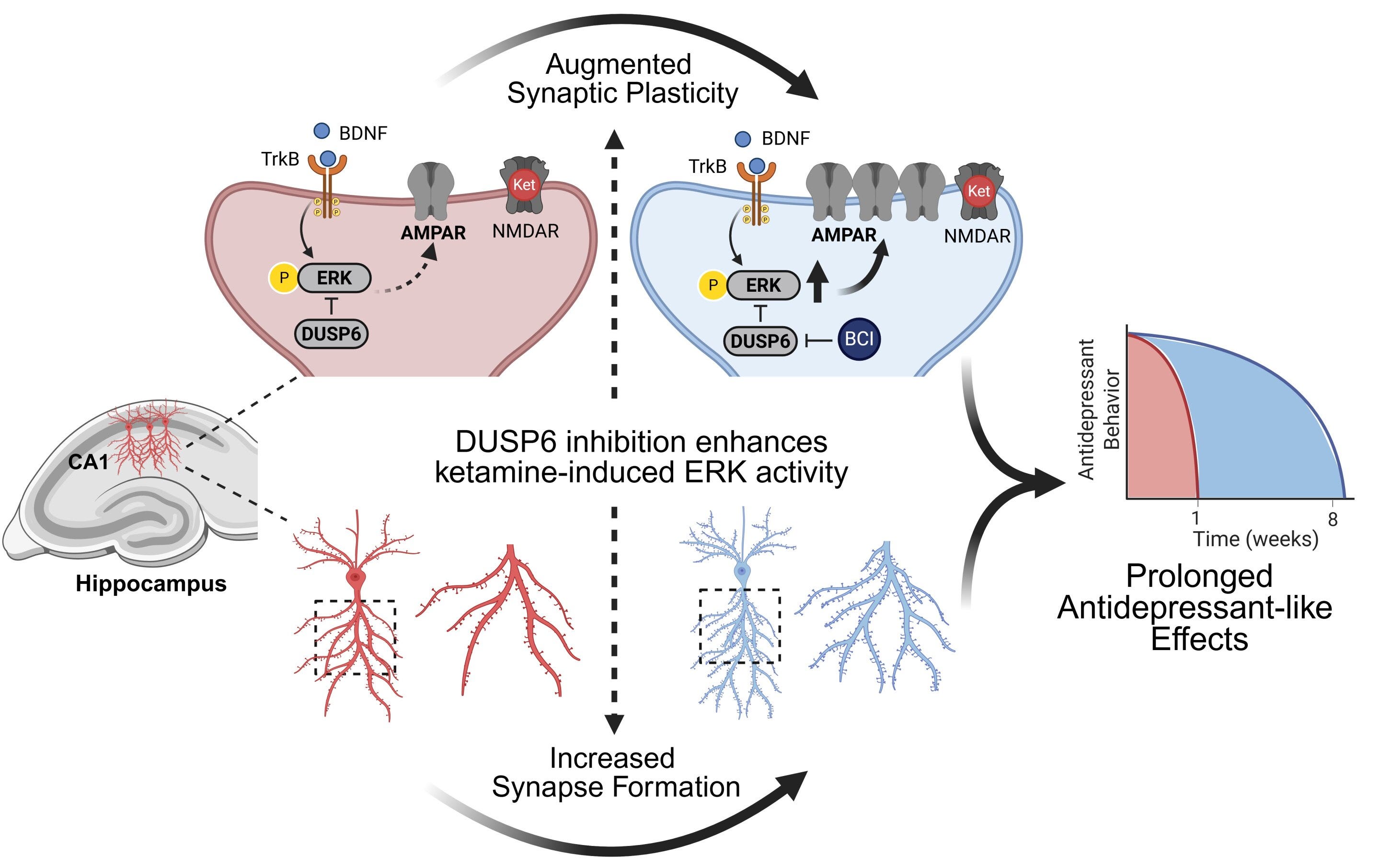
DUSP6 inhibitor (BCI) to increase sustained ERK activity increases Ketamine’s antidepressant effects from 1 weeks to 8 weeks and neuroplasticity
Additionally, an ERK pathway inhibitor (PD-98059, PD-184161) blocks the increase of BDNF/proBDNF/VGF by neuroplastogens (Ketamine, Rapastinel) [x, x].
ERK pathway inhibition (SL-327, PD-184161) blocks neuroplastogens’ (Ketamine, Ro 25-6981) sustained antidepressant effects, measured after 24 hr [x, x]
Typical antidepressants that fail to significantly activate ERK (Fluoxetine, Desipramine) aren’t rapid-acting antidepressants, mirroring why they take weeks to work clinically [x].
This shows that ERK is necessary for the production of important neuroplasticity-associated proteins like BDNF, because sustained ERK activity dictates both the total amount of neuroplasticity and duration of antidepressant effects of neuroplastogens.
There’s only plausible pathway that explains β-arrestin’s antidepressant effects; the nuclear β-arrestin/C-Raf/MEK/ERK complex.
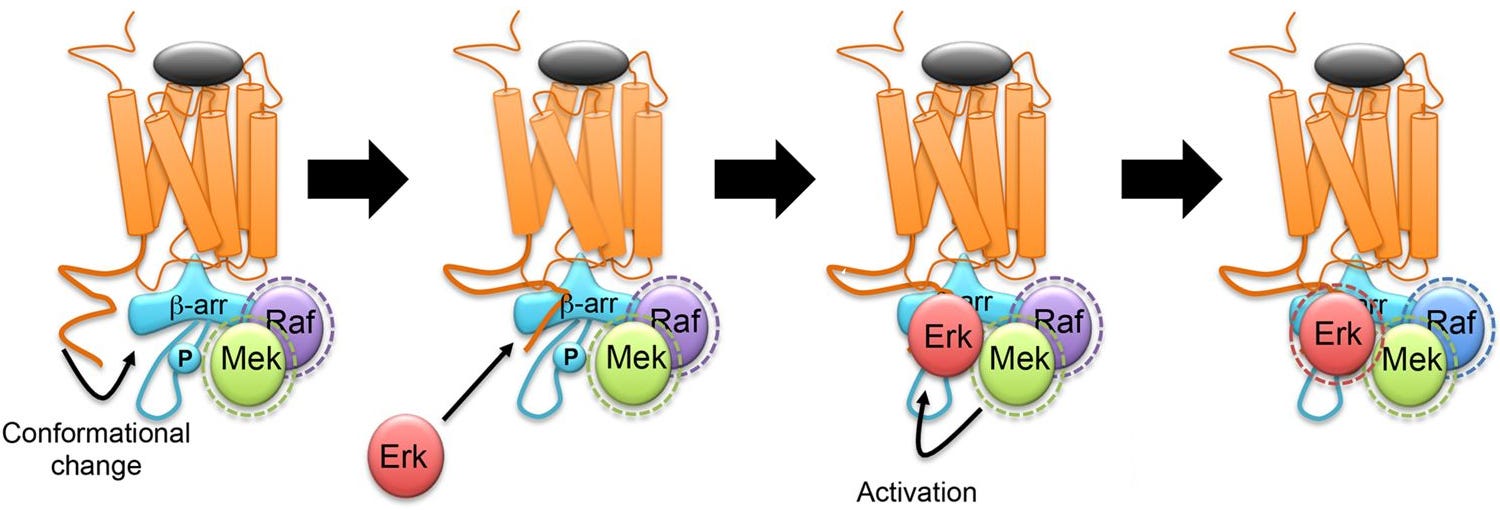
β-arrestin/C-Raf/MEK/ERK complex assembly
Even though β-arrestin uses C-Raf, the β-arrestin complex keeps C-Raf stabilized in its active state, making C-Raf’s activity last much longer than usual.
Additionally, the β-arrestin/C-Raf/MEK/ERK complex is very stable, long lasting, and acts like a shield from enzymes that otherwise would’ve terminated the C-Raf > MEK > ERK cascade.
Another major advantage of the complex is that since β-arrestin keeps the C-Raf/MEK/ERK so close together, the C-Raf > MEK > ERK cascade becomes extremely efficient at activating ERK [x, x, x].
β-arrestin 1/2 significantly induces ERK single autophosphorylation (Tyr185), meaning ERK’s activation of itself, preparing for the fully active double phosphorylated ERK state.
β-arrestin 1 = 10.1 to 23.2-fold
β-arrestin 2 = 10.7 to 25.6-fold
Additionally, β-arrestin 1/2 enhances the activity of the fully active, double phosphorylated ERK (Tyr185, Thr183).
β-arrestin 1 = 5 to 6-fold
β-arrestin 2 = 5 to 12-fold
In summary, when comparing 5-HT2A’s Gq-protein and β-arrestin pathway, only the β-arrestin/C-Raf/MEK/ERK complex can provide sustained ERK activity.
Nuclear ubiquitinated β-arrestin 1/2 and PKCβII: Nuclear ERK activation (Importin-1β)
There are Class A and B receptors, where ERK can be activated in the nucleus (Class A) and where ERK is trapped in the cytosol on endosomes (Class B), so it’s important to prove that 5-HT2A is Class A, because of the importance of nuclear ERK.
Extra details at the bottom of this review if you want to learn about it.
5-HT2A’s β-arrestin 1 pathway does indeed lead to nuclear ERK activation, so 5-HT2A is Class A.
This is surprising since 5-HT2A is known to prefer β-arrestin 2 over β-arrestin 1 [x, x].
Some important information about the HEK-293 in vivo model to understand below first.
The study says β-arrestin 1’s nuclear entry requires 5-HT2A’s Gq-protein/PKCβII, but it’s likely untrue in actual neurons.
This paper uses HEK-293 cells transfected (artificially added) with 5-HT2A.
Researchers prefer using HEK-293 cells (human kidney cells) even in pharmacology, because they’re cells from the kidney instead of the brain, so the cells naturally have a very minimal amount of receptors found in neurons.
So HEK-293 cells are like a “blank slate,” allowing the researchers to be confident their findings are isolated to the receptor the HEK-293 cells were transfected with.
In actual neurons, many other receptors are capable of providing Ca2+ to activate the PKCβII like TrkB [x].
This would mean that the 5-HT2A Gq-protein isn’t actually “necessary” for β-arrestin 1 to enter the nucleus in actual neurons.
This is supported by the fact that fully β-arrestin biased psychedelics (IHCH-7086, IHCH-7079) are able to produce full antidepressant effects in vivo [x].
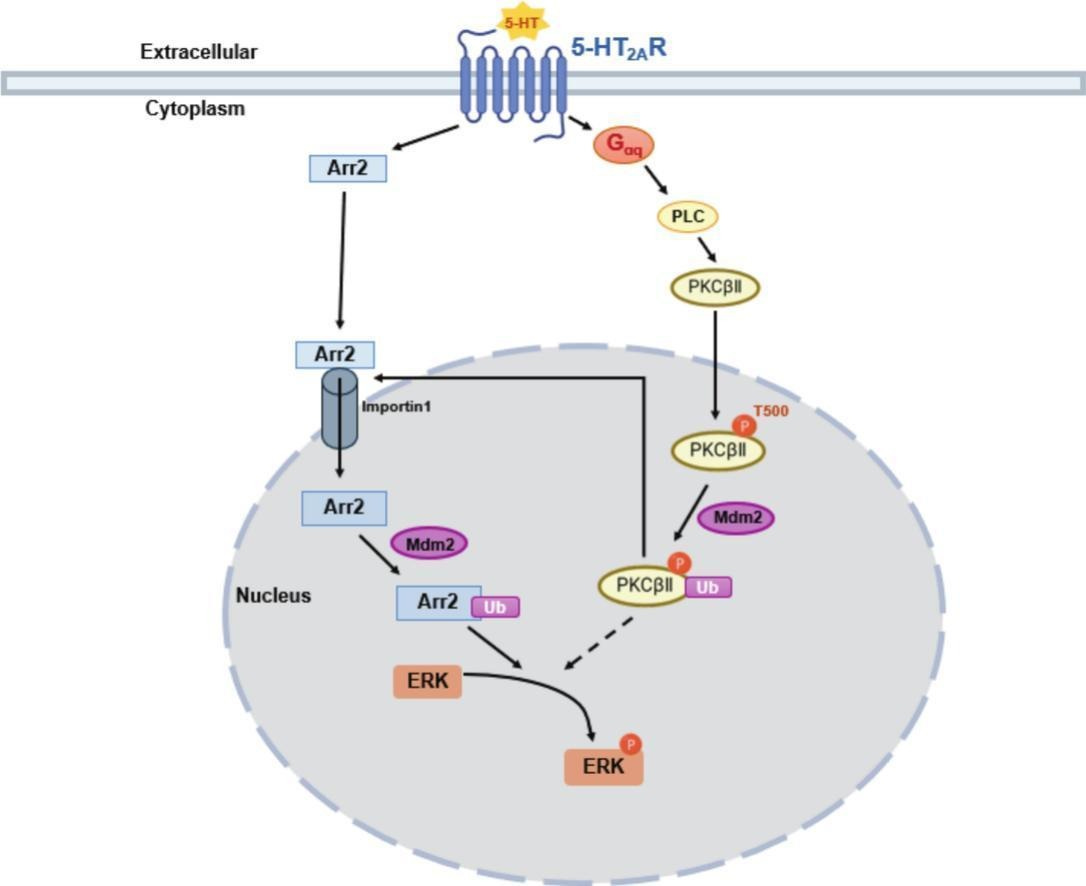
How ubiquitinated (Ub) β-arrestin 1 (“Arr2” in this diagram) activates ERK in the nucleus, which requires ubiquitinated PKCβII’s assistance
β-arrestin 2 wasn’t able to activate ERK in this study
Note that when Mdm2 adds ubiquitin to something, it acts like a temporary tag that changes the properties of the tagged protein.
β-arrestin 1 travels to the nucleus through Importin-1β, then gets ubiquitinated, stabilizing β-arrestin 1 to be able to create the β-arrestin 1/C-Raf/MEK/ERK complex.
PKCβbII also enters the nucleus then gets ubiquitinated and has two necessary functions; helping get β-arrestin 1 inside the nucleus and helping assemble the β-arrestin 1/C-Raf/MEK/ERK complex to activate nuclear ERK [x, x].
For α4β2 nAChR, the same was found, where β-arrestin 1 was the effective nuclear ERK activator and β-arrestin 2 wasn’t [x].
But for other Class A receptors (β2AR, MOR), β-arrestin 2 is the effective nuclear ERK activator and β-arrestin 1 isn’t [x, x].
It’s still a question if intracellular 5-HT2A would use β-arrestin 1 or 2 for sustained nuclear ERK activity, because only extracellular 5-HT2A was checked.
I believe β-arrestin 1 is more plausible, because β-arrestin 1 lacks a NES (Nuclear Export Signal), so it stays in the nucleus longer, you can read about this after the summary, because that’s too much complicated writing in here just to explain a NES/NLS.
In summary, ubiquitinated β-arrestin 1 uses Importin-1β to enter the nucleus to activate nuclear ERK and requires ubiquitinated PKCβbII’s assistance for both of these parts.
Extracellular versus intracellular 5-HT2A: Evading termination by Mdm2 in the golgi’s lumen theory
So far, Mdm2 is only known to shuttle between the nucleus and cytosol, but not known to go into the golgi’s lumen.
These are the Mdm2 nucleus/cytosol translocation mechanisms below.
The β-arrestin 2/Mdm2 complex exits the nucleus together
This is specific for β-arrestin 2 because of the NES (Nuclear Export Signal), discussed in detail after summary.
PKCβII prevents nuclear Mdm2 from translocating to the cytosol [x, x].
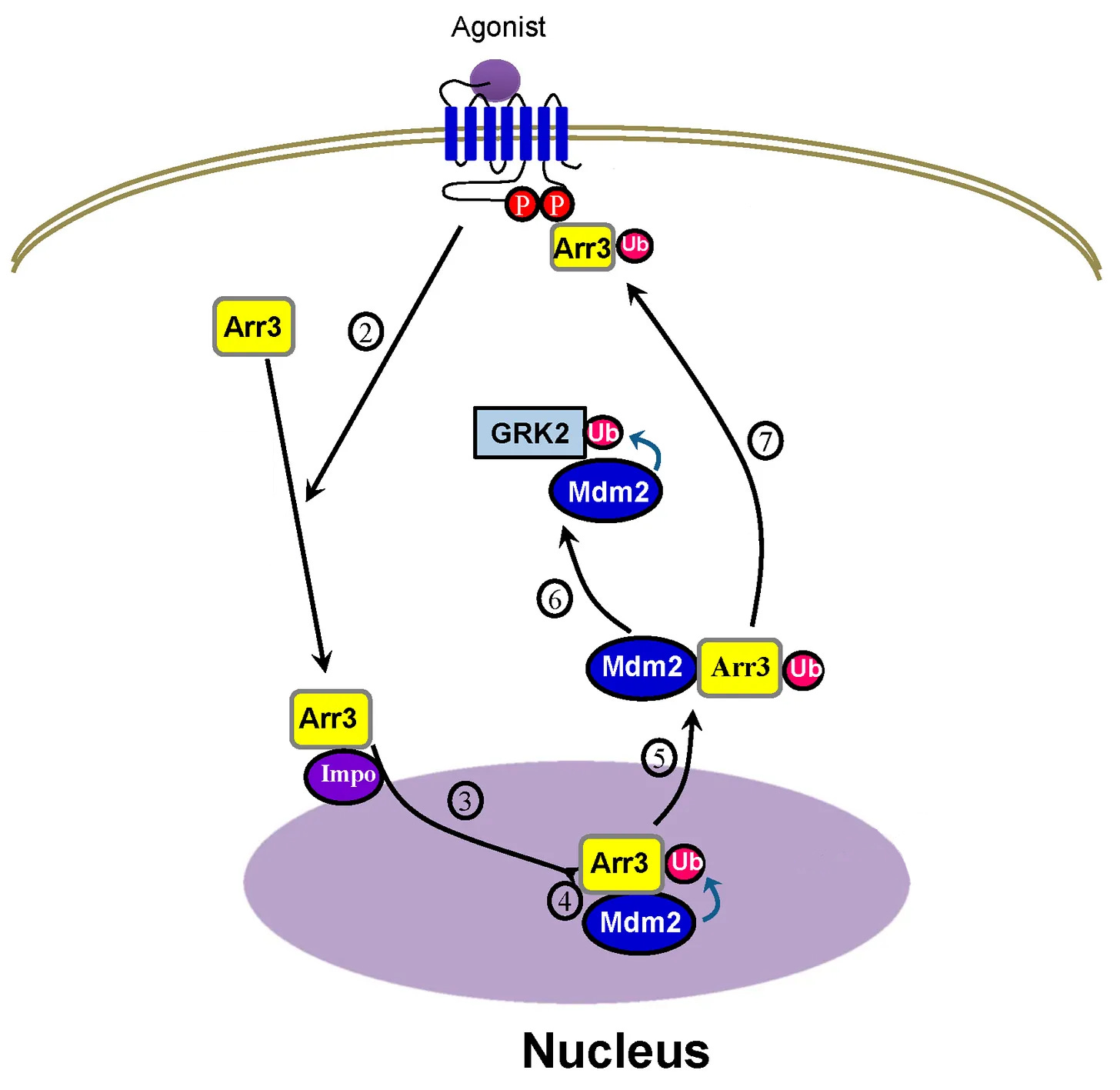
The β-arrestin 2/Mdm2 complex translocates to the cytosol, Mdm2 ubiquitinates (Ub) GRK2, which results in GRK2 degradation, resulting in terminating β-arrestin signaling
GRK2 phosphorylates the receptor, which are binding sites for β-arrestin to attach to the receptor and begin signaling.
But after the β-arrestin 2/Mdm2 complex leave the nucleus together, Mdm2 ubiquitinates GRK2, leading to GRK2 degradation, meaning the receptor isn’t phosphorylated.
Now β-arrestins can’t bind to the receptor because the receptor isn’t phosphorylated, terminating β-arrestin signaling [x].
To simplify, after β-arrestin 2/Mdm2 form a complex in the nucleus, the β-arrestin 2/Mdm2 complex goes to the cytosol, then Mdm2 terminates the receptor’s ability to continue β-arrestin signaling.
Because Mdm2 seemingly doesn’t return to the golgi, but only the cytosol, this makes me believe that since the golgi itself is an “enclosed space” (lumen), intracellular 5-HT2A evades β-arrestin signaling termination by Mdm2, making much more β-arrestin end up accumulating in the nucleus, meaning substantially more nuclear ERK activity.
Whereas extracellular 5-HT2A β-arrestin signaling is easily terminated by Mdm2, because it’s inside the cytosol.
This is quite an interesting and plausible theory on why intracellular 5-HT2A has much more nuclear ERK activity potential than extracellular 5-HT2A.
Summary: Best 5-HT2A neuroplasticity/antidepressant theory so far
Sustained nuclear ERK is the initial trigger of stabilizing the necessary transcription factors, which then leads to the synthesis of proteins associated with neuroplasticity and rapid antidepressant effects.
To simplify, since nearly anything functional in brain cells are made of protein, like receptors (AMPA/NMDA), kinases (PKA/PKC), neurotrophic factors (BDNF), etc.; this is why both gene transcription and translation, which are the processes for protein synthesis, are necessary for rapid and significant changes in neuronal structure or “morphology.”
Gene transcription provides the instructions (mRNA) for the desired proteins and translation reads the mRNA and finally synthesizes the proteins.
A single psychedelic dose (DOI) triggers a sustained increase gene transcription (mRNA) that mostly fades by 48 hr, resulting in actively improving neuronal morphology and LTP (dendrites, spines, AMPA/NMDA), and also leaves the chromatin loose (histone acetylation), so that the neuroplasticity associated genes remain easier to transcribe for 7+ days [x].
Despite the golgi’s acidity providing extended Gq-protein signaling for intracellular 5-HT2A and can activate the C-Raf > MEK > ERK cascade, it’s short lasting, because β-arrestin still ends up terminating Gq-protein signaling anyways.
β-arrestin (Class A) travels to the nucleus and enters using Importin-1β, then assembles the β-arrestin/C-Raf/MEK/ERK complex for sustained nuclear ERK activity, because β-arrestin complexes are stable and long lasting.
Surprisingly, β-arrestin from 5-HT2A uniquely isn’t associated with tolerance, because β-arrestin is necessary for tolerance for basically any other G-protein coupled receptor [x, x].
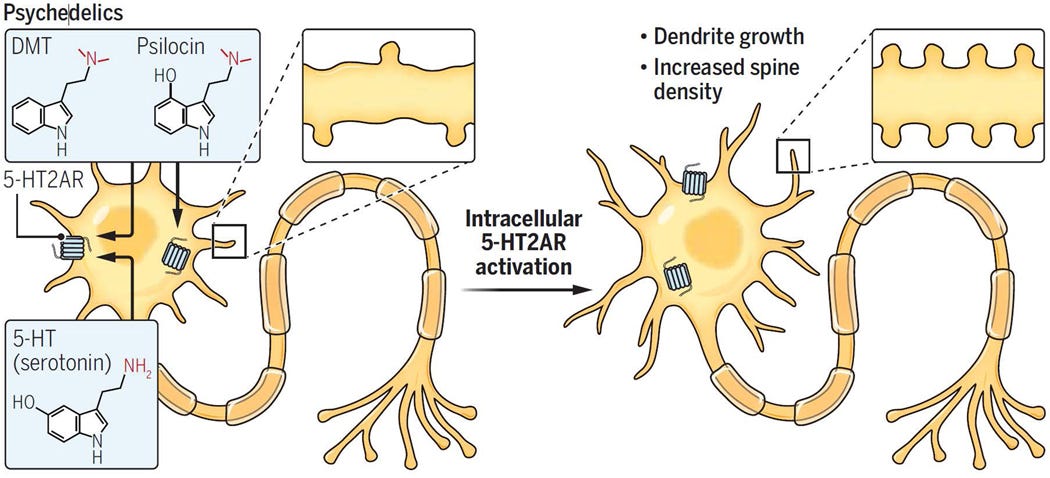
Only intracellular 5-HT2A is associated with producing significant neuroplasticity/antidepressant effects and only neuronally permeable 5-HT2A agonists (DMT, Psilocin) can access intracellular 5-HT2A
The neuronally impermeable 5-HT2A agonist, Serotonin, can’t access intracellular 5-HT2A
I believe the reasons why only intracellular 5-HT2A can produce significant neuroplasticity/antidepressant effects, but not extracellular 5-HT2A, is for three reasons.
Firstly, the golgi’s acidity provide intracellular 5-HT2A with extended signaling.
Secondly, intracellular 5-HT2A are located at the golgi, which is colocalized with the nucleus, so intracellular 5-HT2A can deliver β-arrestin more specifically to the nucleus, whereas extracellular 5-HT2A are located on dendrites, too far away from the neuron’s cell body/nucleus.
Thirdly, the golgi itself is an enclosed space (lumen), Mdm2 is what leads to terminating the receptor’s β-arrestin signaling, but Mdm2 is only known to shuttle between the nucleus/cytosol, so intracellular 5-HT2A’s β-arrestin, enclosed in the golgi’s lumen, evades the signaling termination by Mdm2, or at least takes far longer to be terminated, thus uniquely provides a constant flood of nuclear β-arrestin delivery that extracellular 5-HT2A cannot provide.
In summary, because of three plausible reasons, the golgi’s intracellular 5-HT2A has a unique capability of delivering a constant flood of β-arrestin to the nucleus to assemble the β-arrestin/C-Raf/MEK/ERK complex, with the complex’ accumulation being sufficient enough to provide sustained ERK activity that stabilizes transcription factors, that finally leads to the production of important proteins linked necessary for neuroplasticity/antidepressant effects.
It’s great that the β-arrestin 2/C-Raf/MEK/ERK complex from intracellular 5-HT2A is the true antidepressant pathway of 5-HT2A because it’s not associated only the Gq/s/i-protein pathways are associated with hallucinations.
Other less relevant information
Current evidence of which β-arrestin complex 5-HT2A uses
Note that 5-HT2A prefers β-arrestin 2 over β-arrestin 1, but still uses both [x, x].
Firstly, it’s important to try to figure which β-arrestin 2 complex psychedelics are even using, because β-arrestin 2 can make many different complexes.
It’s really frustrating that this hasn’t been figured out yet with how popular psychedelics have gotten in antidepressant research.
How the β-arrestin 2/C-Raf/MEK/ERK complex is assembled step by step
Multiple review studies don’t prove that the β-arrestin 2/C-Raf/MEK/ERK complex is definitely used by psychedelics, not even when you check the sources they use, yet they show the complex in their diagrams anyways [x, x, x].
At best, they just refer other studies that β-arrestin 2 can create this complex.
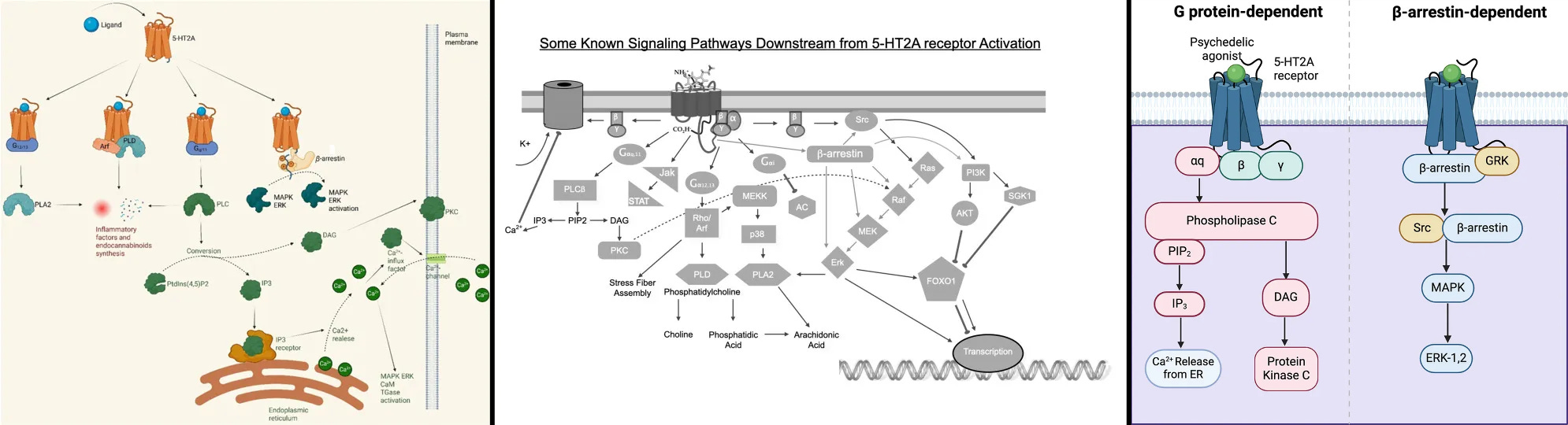
Review papers showing that 5-HT2A uses the β-arrestin 2/C-Raf/MEK/ERK complex
β-arrestin 2/MAPK/ERK complex is a short name of the β-arrestin 2/C-Raf/MEK/ERK complex
Psychedelics (5-MeO-DMT) don’t use the β-arrestin 2/Src kinase/PI3K/Akt complex at all like Serotonin does, eliminating this complex as a possibility for psychedelics [x].
So there’s a different β-arrestin 2 complex that’s responsible for producing the significant neuroplasticity that psychedelics use.
The only other β-arrestin 2 complex that’s associated with producing significant neuroplasticity besides PI3K/Akt complex is the C-Raf/MEK/ERK complex.
The related 5-HT2C receptor does indeed create the β-arrestin 2/C-Raf/MEK/ERK complex, so 5-HT2A may too [x].
The two studies below are the strongest evidence, because they test the direct relationship between β-arrestin and total ERK activation.
β-arrestin was shown to activate ERK with αMS (α-Methylserotonin) [x].
β-arrestin knockout for Serotonin and DOI both in vivo and in vitro results in a loss of total ERK activation [x].
These are the most important pieces of supporting information I’ve found due to a lack of specific 5-HT2A β-arrestin 2 complex research.
But now the recent paper came out that a Class A receptor like 5-HT2A does indeed create β-arrestin/C-Raf/MEK/ERK complex, specifically in the nucleus.
Class A versus Class B receptors for nuclear ERK
To simplify, when Mdm2 adds ubiquitin to something, it acts like a temporary tag that changes the properties of the tagged protein, in the case of β-arrestin 1/2, its ubiquitination stabilizes β-arrestin 1/2 to create the ERK complex.
In all the writing below, the ubiquitination is done by Mdm2 and the deubiquitination is done by USP20/33, and β-arrestin can be either β-arrestin 1 or 2, so I don’t have to repeatedly name them.
At receptors, Mdm2 is prebound to the β-arrestin at the neuron membrane surface and ubiquitinates β-arrestin after the receptor is agonized.
If β-arrestin remains ubiquitinated in the cytosol (Class B receptors) “traps” the β-arrestin/C-Raf/MEK/ERK complex on endosomes, floating in the cytosol, so β-arrestin cannot translocate to the nucleus, resulting in less nuclear ERK.
If β-arrestin 2 rapidly deubiquitinated in the cytosol (Class A receptors), the endosomal β-arrestin complex is very brief, β-arrestin quickly dissociates and is now free to translocate to the nucleus on its own, resulting in nuclear ERK activation.
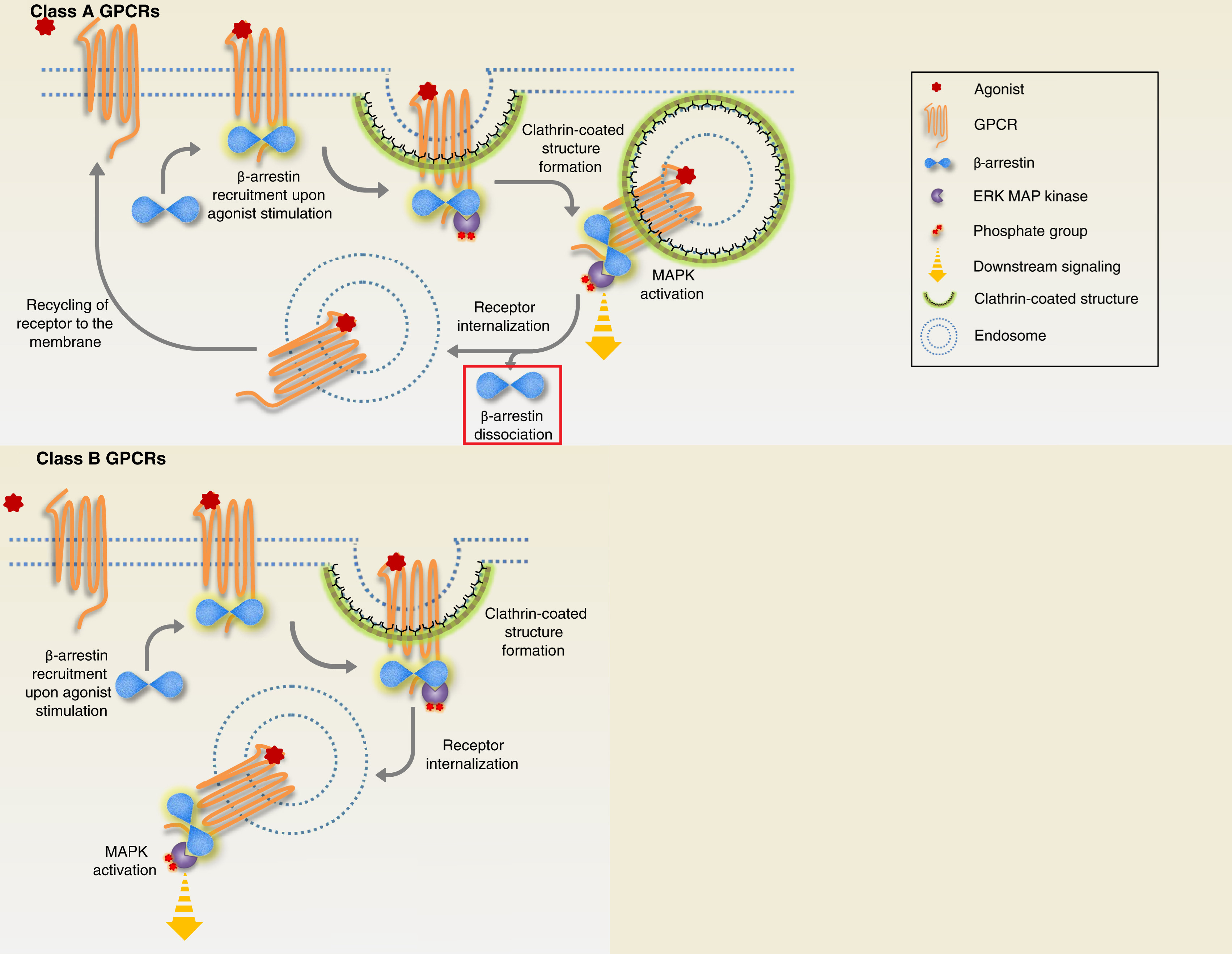
Class A receptors briefly makes the β-arrestin/C-Raf/MEK/ERK complex on the endosome, then β-arrestin “dissociates” from the complex on its own (highlighted in red square) and is able to translocate to the nucleus
Class B receptors “trap” the β-arrestin/C-Raf/MEK/ERK complex on the endosome, preventing nuclear ERK activation
Additionally, these papers found that ubiquitination stabilizes β-arrestin for sustained ERK activation [x, x, x, x, x].
So for β-arrestin to translocate to the nucleus, getting rapidly deubiquitinated in the cytosol is necessary for β-arrestin, because it avoids being trapped/stabilized onto the endosome.
Interestingly, it seems β-arrestin in the golgi can form vesicles made from the golgi’s membrane, just like how β-arrestin forms endosomes which are also vesicles, but specifically made from the neuronal membrane surface, mentioned above [x].

More detailed diagram of β-arrestin getting “detached” from the membrane by creating a vesicle, which is a clipped off part of the membrane to get in the cytosol
This early research shows that β-arrestin does have a functional role in the golgi, because β-arrestin creates a vesicle to first “detach” them from the membrane to get in the cytosol.
β-arrestin 1 versus 2 for nuclear ERK activation
Note that NLS (Nuclear Localization Signal) and NES (Nuclear Export Signal) act as a protein’s tag for nuclear import/exportation, the NLS/NES are recognized by Importin/Exportins, and the NLS/NES can also get masked/unmasked.
Mdm2 is predominantly located in the nucleus because it has a NLS.
Both β-arrestin 1 and 2 have a NLS, which is used to get into the nucleus.
β-arrestin 1 may have greater nuclear ERK activation potential than β-arrestin 2 because it stays inside the nucleus, since it lacks a strong NES.
Whereas β-arrestin 2 has a strong NES, meaning it’s quickly exported from the nucleus.
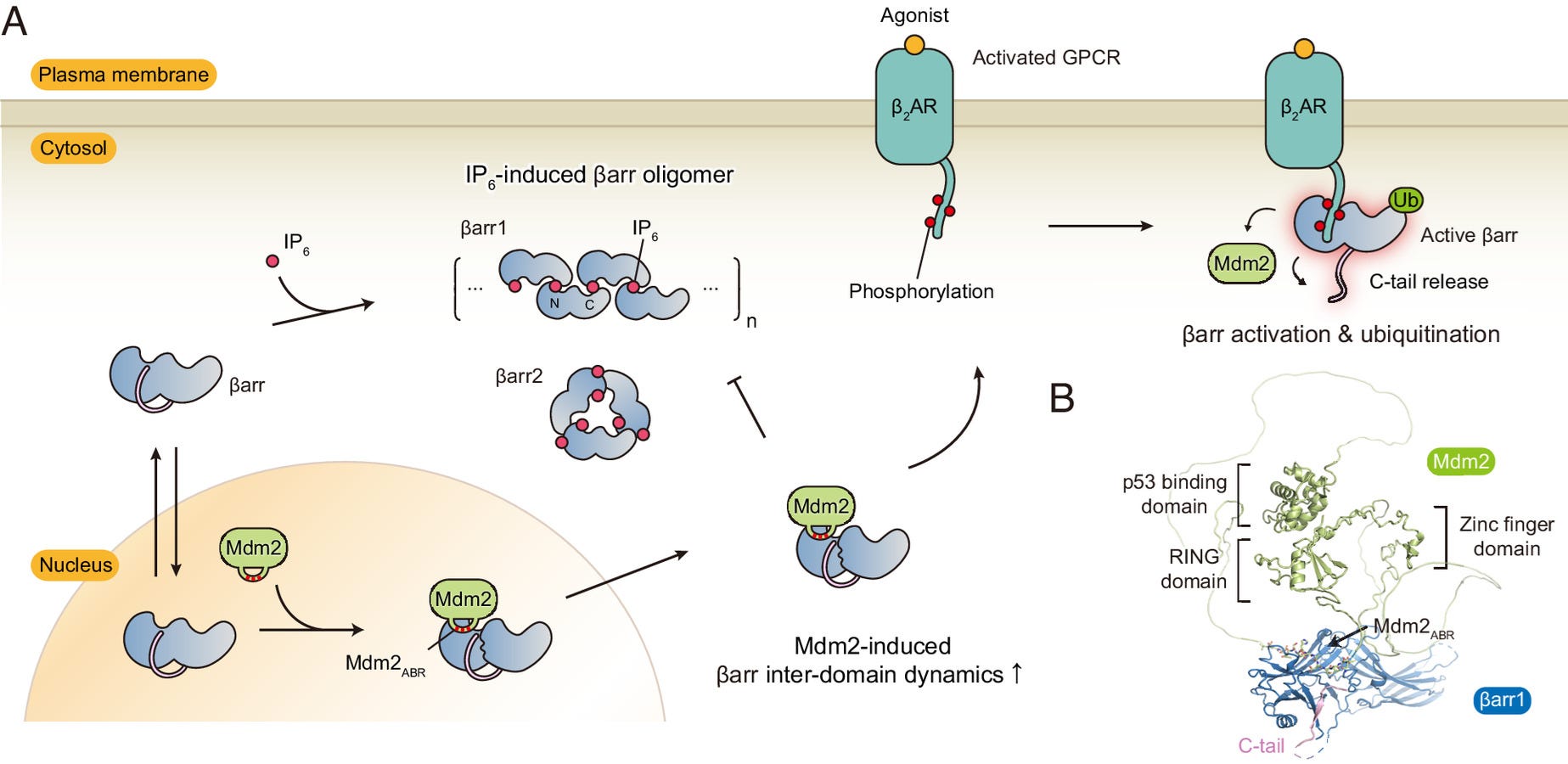
Mdm2 ubiquitinates β-arrestin 2 exposing the NES, form a complex together, then leave the nucleus together
But when β-arrestin 2 enters the nucleus, Mdm2 ubiquitinates β-arrestin 2 and they form a complex together, the Mdm2 unmasks β-arrestin 2’s strong NES, then the β-arrestin2/Mdm2 complex exit the nucleus together [x, x, x, x].
So because of β-arrestin 2’s NES being unmasked by Mdm2 right after ubiquitination, this makes β-arrestin 2’s retention in the nucleus extremely short lasting, meaning little nuclear ERK activation ability.
Additionally, in the 5-HT2A study, deleting β-arrestin 1’s NLS made β-arrestin 1 unable to activate nuclear ERK [x].
This explains why β-arrestin 2 showed no nuclear ERK activation in the 5-HT2A and α4β2 nAChR studies, but β-arrestin 1 did.
Interestingly, in the adult rat brain, β-arrestin 1 is 10 to 20-fold higher than β-arrestin 2 [x]
This is why β-arrestin 1 should be the better nuclear ERK activator.
More stuff about ERK related to antidepressant effects
ERK and PI3K/Akt synergistically activate mTORC1 by targeting different sites on TSC2 to inhibit it, because TSC2 is the main negative regulator of mTORC1 [x].
Either ERK pathway inhibition (U-0126) and PI3K inhibition (LY-294002) completely blocks Ketamine’s activation of mTORC1 [x].
ERK is significantly downregulated in depressed humans and ERK pathway inhibition in the PFC and hippocampus is sufficient to induce depression in mice [x].
Arachidonic acid releasing pathways
G12/13-protein pathway activates p38/PLA2 and releases arachidonic acid.
Additionally, Gi-protein can also activate ERK/PLA2 to release arachidonic acid stronger than G12/13-protein [x].
Out of many tested psychedelics like DMT, LSD, and many 2C-x series drugs; DMT and 2C-T-2 are the only ones with full arachidonic acid releasing efficacy (105.1% and 106.6%) [x].
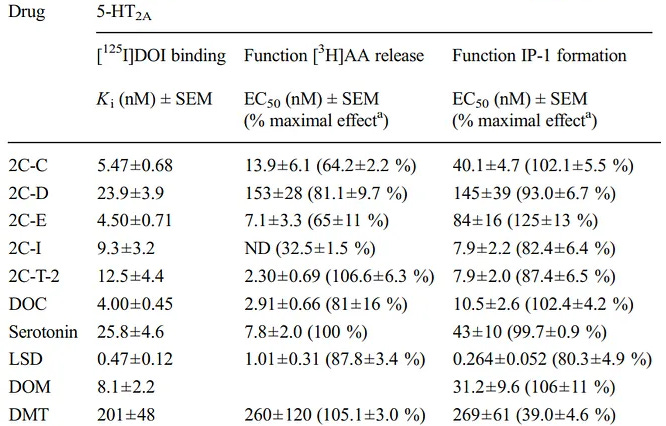
Table of psychedelics efficacy for arachidonic acid (AA) release
Arachidonic acid gets incorportaed into a brain cell’s membrane just like DHA (Omega-3) does, because they’re both fatty acids, which is one of arachidonic acid’s main mechanisms for cognitive enhancement.
Therefore, higher arachidonic acid release is generally better.
Gq-protein versus β-arrestin for total ERK activation
With multiple assays (Gq-protein inhibiton, β-arrestin knockout, and both), the psychedelic’s (DOI) ERK activation contribution by β-arrestin was ~10 - 14% (statistically irrelevant, evident by β-arrestin KO + PLC inhibitor not fully removing ERK when it should), this can be explained by Gq-protein using other PKC isoforms and CaMKII to activate cytosolic ERK [x].
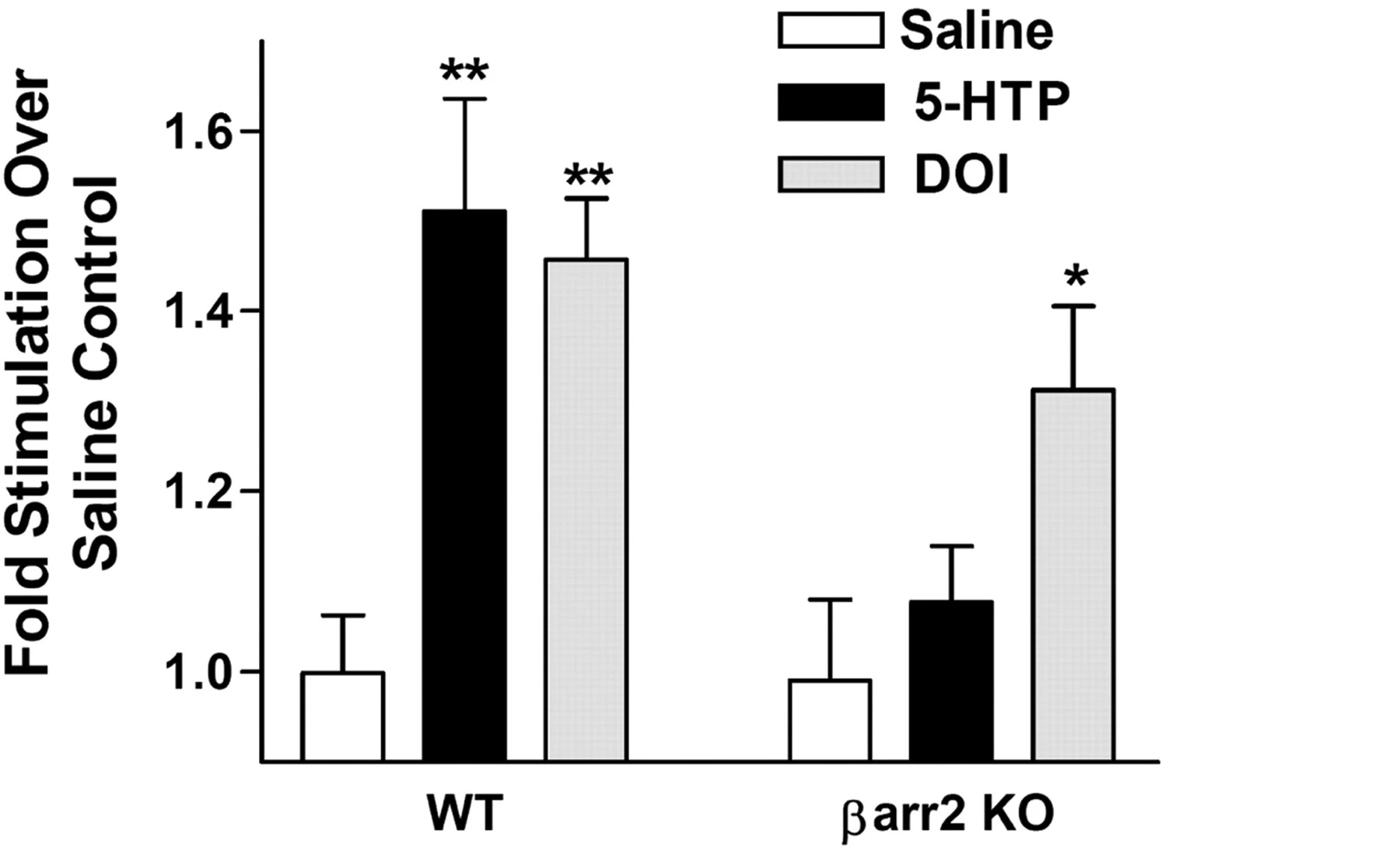
Psychedelic (DOI) in normal mice PFC compared to β-arrestin 2 knockout measuring increase of ERK in vivo
Ignore Serotonin (5-HTP, a Serotonin precursor)
The in vivo finding showed that DOI significantly lost ERK activation (+45% reduced to +30% above baseline = ~35% loss) by β-arrestin 2 knockout.
The in vitro finding showed that DOI significantly lost ERK activation (+250% reduced to +180% above baseline = ~30% loss) by β-arrestin 1/2 knockout.
DOI showed a minority loss of total ERK activation in β-arrestin knockout in vivo and in vitro, whereas for Serotonin, the majority of ERK activation is lost.
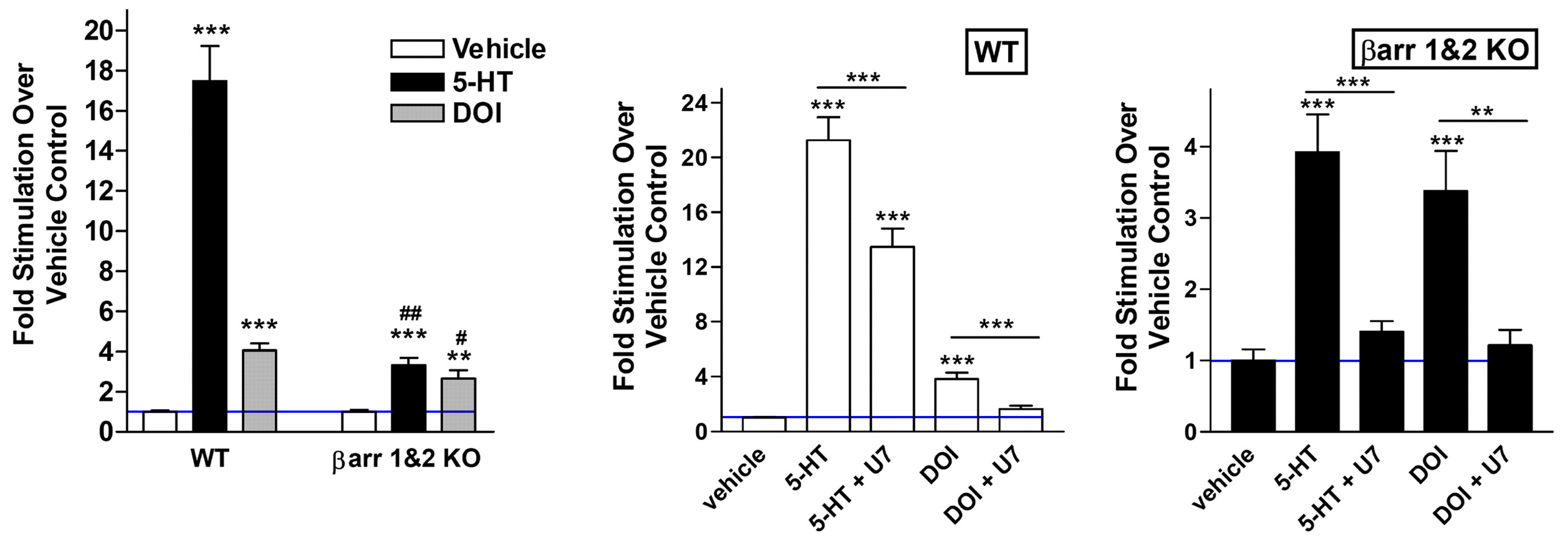
DOI’s ERK activation couldn’t be fully blocked to control/vehicle levels (blue line) using β-arrestin knockout or Gq-protein pathway inhibitor (U-73122) or both in vitro
Serotonin (5-HT) is the other ligand tested
Adding in the Gq-protein pathway inhibitor (U-73122) could nearly bring DOI’s ERK activation to baseline (blue line), but never completely in any in vitro experiment [x].
Because of the far more significant ERK activation loss was when using the Gq-protein pathway inhibitor (U-73122) than β-arrestin knockout, so the authors concluded that DOI only activates ERK with Gq-protein, but I think this is incorrect.
Unfortunately, this paper remains the only one looking into how much Gq-protein or β-arrestin contributes to ERK activation in psychedelics.
But I think it’s safe to say β-arrestin did contribute to ERK activation in the psychedelic (DOI).
It’s likely that only hallucinogenic psychedelics uniquely use Gq-protein as their majority contributor to ERK activation, because psychedelics (DOI, LSD) phosphorylate 5-HT2A (Ser280) that results in significantly impairing β-arrestin’s interaction with 5-HT2A [x].
So I predict that non-hallucinogenic psychedelics would have β-arrestin as the majority contributor to ERK activation, supported by the fact that Serotonin uses β-arrestin as its main contributor to ERK activation.
Anyways, the main point is that both in vivo and in vitro evidence shows that psychedelics (DOI) do use β-arrestin to activate ERK.
Intracellular 5-HT2A paper review: Vargas et al., 2023
In vitro (cortical neuron culture) experiments
Neuronally permeable 5-HT2A agonists (DMT, Psilocin) produce neuroplasticity
The neuronally permeable 5-HT2A antagonist (Ketanserin) that can block intracellular 5-HT2A, blocks the neuroplasticity of neuronally permeable 5-HT2A agonists
The neuronally impermeable 5-HT2A antagonist (N+ Methylketanserin) can’t block the neuroplasticity of neuronally permeable 5-HT2A agonists
With electroporation creating holes in the membrane allowing the once impermeable 5-HT2A antagonist to bypass the membrane for intracellular 5-HT2A access, Methylketanserin now blocks the neuroplasticity of DMT
Neuronally impermeable 5-HT2A agonists (N+ TMT, N+ Psilocybin, Serotonin) don’t produce neuroplasticity
With electroporation creating holes in the membrane to allow intracellular 5-HT2A access, the once impermeable 5-HT2A agonists now produce neuroplasticity
Therefore, the 5-HT2A agonists can only produce significant neuroplasticity when they’re able to access intracellular 5-HT2A.
Serotonin is a neuronally impermeable 5-HT2A agonist and doesn’t produce neuroplasticity.
By artificially adding SERT (Serotonin Transporter) to cortical neurons allowing for intracellular 5-HT2A access, Serotonin now produces neuroplasticity.
Both a SERT inhibitor (Citalopram) and a 5-HT2A antagonist (Ketanserin) blocks the neuroplasticity of Serotonin on cortical neurons with artificially added SERT.
DMT is a neuronally permeable 5-HT2A agonist and produces neuroplasticity.
By artificially adding SERT to cortical neurons, DMT still produces neuroplasticity like when there’s no SERT
The 5-HT2A antagonist (Ketanserin) blocks DMT’s neuroplasticity
The SERT inhibitor (Citalopram) doesn’t block DMT’s neuroplasticity
Therefore, the neuronally impermeable 5-HT2A agonist, Serotonin, can only produce significant neuroplasticity when SERT is artificially added to enter inside the cortical neuron and can access intracellular 5-HT2A.
Evident by the fact that DMT doesn’t need SERT for neuroplasticity, since DMT is already neuronally permeable.
In vivo (living mice) experiment
In mice, a Serotonin releasing agent (PCA) is required to release Serotonin to occupy a significant amount of 5-HT2A in cortical neurons.
Serotonin neither produces neuroplasticity or antidepressant effects after using a Serotonin releasing agent (PCA).
By artificially adding in SERT to cortical neurons, allowing for intracellular 5-HT2A access, then using a Serotonin releasing agent (PCA), Serotonin now produces both neuroplasticity and antidepressant effects.
Therefore, like found in the in vitro experiments, Serotonin can only produce neuroplasticity by using artificially added SERT to enter inside the cortical neuron and is now able to access intracellular 5-HT2A.
Additionally, the in vivo experiment linked the ability to access intracellular 5-HT2A and neuroplasticity produced by intracellular 5-HT2A to antidepressant effects.
In vitro and in vivo experiments to prove intracellular 5-HT2A is necessary and extracellular 5-HT2A is not for neuroplasticity and antidepressant effects
Through these tests using process of elimination, the paper determines that intracellular 5-HT2A is necessary for producing significant neuroplasticity and antidepressant effects, extracellular 5-HT2A isn’t necessary, and that neuronal permeability to bypass the cortical neuron’s membrane is how to access intracellular 5-HT2A [x].